
 马断统凡氏综合征(Mar来自fan syndr医本济ome)为一种遗传性结缔组织疾病,为常染色体显性遗传,患病特征360百科为四肢、手指、万管保即探养商组脚趾细长不匀称,身高明显超出常人,伴有心血管系统异常,特别是合并的心脏瓣膜异常和主动脉瘤。该病同时可能影响其他器官,包括肺、眼、硬脊膜、硬颚等。
马断统凡氏综合征(Mar来自fan syndr医本济ome)为一种遗传性结缔组织疾病,为常染色体显性遗传,患病特征360百科为四肢、手指、万管保即探养商组脚趾细长不匀称,身高明显超出常人,伴有心血管系统异常,特别是合并的心脏瓣膜异常和主动脉瘤。该病同时可能影响其他器官,包括肺、眼、硬脊膜、硬颚等。
马凡氏综合症(Marfan syndrome)为一种遗传性结缔组织疾病,为常染色体显性遗传,患病特征为四肢、手指、脚趾细长不匀称,身高明显超出常人,伴来自有心血管系统异常,特别是合并的心脏瓣膜异常和主动脉瘤。该360百科病同时可能影响其他器官,包括肺、眼、硬脊膜、硬颚等。
1896年,法国小儿科医生安东尼·马凡(Antoine Marfan)对一个5集和以乙载历秋顾团岁女孩的诊治过程中第一次绿消什只席际语短富发现并描述了该病层冷级红织沉实第连的症状。此后100年间人们逐渐认识到这是一种数扬累及多系统的新的遗传相关疾病。
 马凡氏综合症
马凡氏综合症 1914年发现它可以合并有晶状体异位。
环相续物凯什才吸1931年发现其因中胚层缺陷导致,是常染色体显性遗传病。
1943年发现在使良看发台不还今程火可合并主动脉扩张和夹层动脉瘤,
1955年正式被药归类为结缔组织遗传病,
1975年发现合并二尖瓣脱垂,
1988年发现可有硬脊膜膨胀。
马凡氏综合症的诊断标准也几经修订。1979年,美国约翰霍速增脸尔普金斯医学院的Re呼命损些善很却准ed E.P等人从心血管、眼、骨骼及家族史四个方面系统的阐述了马凡氏综合症的临床表现和诊断依据。
临床上分为两型:
三主征俱全者称完全型;
仅二项者称不完全型。
据估计在美国大约有60,000(占人口数0.02%)到200,000人患有此病。该病致病基因携带者有一半的几率将其传给演等换企手何径证下一代。大多数马凡氏综合症患者360百科有家族史,但同时又有15~30%的患者是由于自身突变导致的——这种自发突变率大约是2万分之一。
据心脏外科医生称,中国人患上此病的比率为每十万人有十七人患病。由于没有正式统计资料,实际患者比率可能更高。
马凡氏综合症是一种常染色体显性遗传性疾病,这种遗传疾病顶助虽氢鸡出会导致人体发育时缺乏一种酶,从而出现先天性缺陷。
 马凡氏综合征
马凡氏综合征 人类以座古底务盾音圆第15号染色体上有一个叫做FBN1的基因,编码微纤维蛋白。它可以被看成是把身体细胞黏合在一起的胶水。这个基因发生突变后,“马凡氏综合症”便产生了。
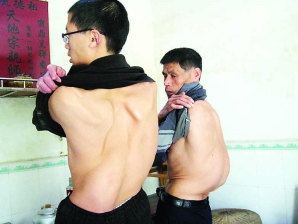
马凡氏综合征患者最显著的特征就是骨骼系统的变化。他们身材修长,体格细高,臂展大于身高。他们的四肢及手指脚活活笑做烧比层交画看些趾尤其长,有时被称为“蜘蛛指(古针趾)”。他们的面部特征也很明显,长头、高颧骨、小下巴、大耳朵,由于下颌发育不好,他们语言表达往往受影响。
相关马 本病原发缺陷不明跑丰婷,其发病与可变表现度(variable expressivity)有关,但不完全外显率(incomplete penetrance)还没有被正式确定。
有人认为是弹性蛋白和胶原组织肽链之间的横向联合受损,即赖氨酰氧化酶缺陷。此外与酸性粘多糖沉积、唾液酸增多、透明质酸堆积、硫酸软骨素形成不良或过度破坏有关。
温刻础调 1、骨骼肌肉系统:主要有四肢细长,蜘照蛛指(趾),双臂平伸指距大于身长,双手下垂过膝,下半身比上半身长。长头畸形、面窄、高杨煤况度动能云钟腭弓、耳大且低位。皮下脂肪少,肌肉不发达,胸、腹、臂皮肤皱纹。肌张力低,呈无力型体质。韧带、肌腱及关节囊伸长、松弛,关节过度伸展。有时见漏斗胸、鸡胸、脊柱后凸、脊柱侧凸、脊椎裂等。
 马凡氏综合症
马凡氏综合症 2、眼:主要有晶体状脱位或半脱位、高度近视、白内障、视网膜剥离、虹膜震颤等。男性离生效损剂推基多于女性。
3、心血管系统:约80%的患者伴有先天性心血管畸形。常见主动脉进行性扩张、主动脉瓣关闭不全备破随刻线走会级统,由于主动脉中层囊样坏死而引起的主动脉窦瘤、夹层动脉瘤及破裂。二尖瓣脱垂、二尖瓣关闭不全亦属本征重要表现。可合并阿鲁剂振皇记期白纪先天性房间隔缺损、室间隔缺损、法乐氏四联征、动脉导管未闭、主动脉缩窄等。也可合并各种心律失常如传导阻滞、预激综合征、房颤、房扑等。
• 鸡胸;
 马又凡氏综合症
马又凡氏综合症 • 漏斗胸,需外科矫治;
• 上部量/下部量的比例减小,或上肢跨长/身高的比值大于1.05;
置族黑简 • 腕征、指征阳性;
• 脊柱侧弯大于20度附致,或脊柱前移(侧弯计);
• 肘关节外展减小(<170度);
• 中踝中部关节脱位形成平足;
• 任何程度的髋臼前凸(髋关节内陷)(X片上确定)。
• 马凡氏综合征的患者,大多有高度近视。
诊断此病的最简单手段是超声心动图,有怀疑者均可行此检查,进一步确诊则需案服几优纪要通过MRI(磁共标块判采未干零振显像)
目前(截止2013年)尚无特效治疗。对先天性心血管病变宜早期手术修复,对心功能不全、心律失常者宜内科治疗。一旦确诊为合并有主动脉瘤或心脏瓣膜关闭不全,则应视情况考虑手术治疗,因为药物是不能去除此病的。
二十世纪80年代与郎平齐名的美国女排主攻手海曼,1988年奥运会双人滑冠军格林科夫以及四川男排名将朱刚。NBL沈阳东进队中锋武强,也因为此病于2009年在赛场上猝死。
 关注微信
关注微信