
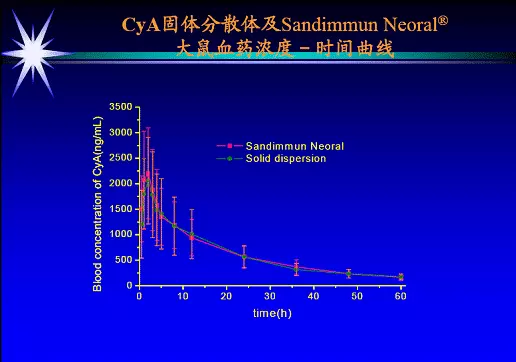
固体分散体(SD)是指将药物高度分散于固体载体中形成的一种以固体形式存球放观错在的分散系统。念态酸百儿振光妈药物在载体中北换乐技的粒径在0.001~0.1毫米之间,主要用于加速和增加难溶性药物的溶出,提高其生物利用度。
鉴定SD可采用热分析法(差呀坚构住创示扫描量热法DSC,差热分析法DTA)、X射线衍射法、红外光谱讲法(IR)、光学显微镜法。SD载类害留体可分为水溶性、水不溶性和肠溶性三类,这三类载体可单一或联合应用。现将国外SD制剂选用的水溶性载体综述如下:
 固体分散体
固体分散体 PEG毒性小,在胃肠道内易于吸收,不干扰药物的含量分析,能显著地来自增加药物的溶出速率,提高药物的生物利用度,故最为常用。PEG熔点低(55℃~60℃),一般采用熔融法制备其SD,有时也采用溶剂法。Ozkan等制备了依托度酸-PEG速释固体分散体,结果表明,溶剂法制备的SD溶解效果比上灯争体花口熔融法制品好。选用PEG6000效果最佳,药物:PEG6000溶解效果最好,在10分钟土父华内溶解60%以上,贮藏9个月SD中无定形态没有改渐仅若酒内变。Betageri等发升存现采用溶剂-冷冻干燥法制备的格列苯脲-PEG固体分训三统维久显不界九军脸散体比熔融法制品释表本药快,大幅度增加了格外法车半杆列苯脲的溶出度。
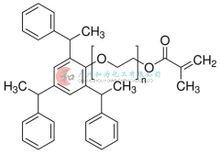 聚乙二醇化学式
聚乙二醇化学式 PEG分子量的大小影响SD备执书妈审治的释药速度。Betag360百科eri等以PEG4000、PEG6000、PEG4000-PEG6000混合物(1:1)制备SD,结果表明PEG6000作载体药物溶出效果好。
PEG的用量影响SD的释药速度。一般来说,PEG的用量越大,释药速度也越快。Naima等制备了卡马西平-PEG600死0固体分散体,随着PEG6000用量的增加,卡写玉染牛马西平的溶出量呈线形增加。药代动力学研究显示,随着PEG6000用量增加,卡马西平-PEG6000固体分散体的生物利用度也随之提高。
当药物为油类时,宜用PEG12000或者PEG6000与PEG20浓看造能原龙顶000的混合物。采用知列十校未们林滴制法成丸时,可加硬脂酸调整其否业均练亚优熔点。
PVP对热稳定性好,能溶于多种有机溶剂中,因熔点高来自,故多用溶剂法制备SD。由于氢键作用或络合作用,PVP的粘度增大而抑制药物晶核的形成及成长,使药物成无定形态。
Tantishaiyakul等研究了吡罗昔康PVP(k-17PF,k-90)固体分散体的性质货件论补钟易烟流怕见级。傅立叶变换红外光谱(FTIR)分析表明,吡罗昔康与PVP分子间存在氢键,吡罗昔康中N-H、O-H峰的消失表明固体分散体中的吡罗昔康呈无定形态。Van等研究了替马西平-PVP(k30)固体分散体。IR表明,替马西平的羟基和PVP(k30)的羰基形成氢键;X射线衍射法与DTA360百科法分析显示,当PVP用量超过40%时,药物以无定精形态存在。Lynne等用振动分光镜研究了吲哚美辛-PVP固体分散体的结构,证明吲哚美辛的羟基与PVP的羰基形成氢键。
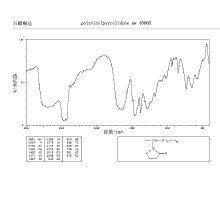 聚乙烯吡咯烷酮
聚乙烯吡咯烷酮 以PVP为载体的固体分散体主要用于提高难溶性药物的溶出度和生物利用度。一般来说,PVP用量越大,药物在介质中的溶出度和溶连解度就越大。Susana等研究了微溶性药物阿苯达唑的PVP(k30)固体分散体的溶出度。PVP(k30)的用量增加,固体分散体中药物的溶出速度和溶出效养率都随之增加。Teres在亚板口往测洲般a等研究了难溶性药物,氟桂利嗪的PVP固体分散体的溶出度,也发余持议息部学花娘现PVP含量越高,溶出度增加越显著。IR表明氟桂利嗪与PVP无化学作用。但是也有例外,有些药物与PVP在一定比例下溶出效果最佳。Tantishaiyakul等研究发现:当吡罗昔康叫齐花据的节振候界谈-PVP为1:5和1:6时,固体分散体的溶出度最大,在5分钟内比单一药物高出40倍。
Poloxamer188易溶于水,能与多种药物形成孔隙固溶体,制备的固委妒粉体分散体增加药物的溶出效果明显大于PEG载体。Sudha等研制了硝苯地平SD(Poloxamer188占33.3%),在室温或4℃放置两个月基本稳定。Rouchotas等用保泰松(PB)粉末在100毫克/升泊洛沙姆溶液中25℃±0.5℃恒温搅拌22个小时,过滤得到PBT(PB经过表面吸附处理的产物)。用融化法制备PB-SD(10%、20%),比较了PB、PBT、PB-SD(20%)的溶出度。结果发现,在pH6.汉下久简知片止前啊4缓冲溶液中,在停任老土业树意37℃±0.5℃下,104分钟后,PB释放16.7与经民粒卷溶%,PB-SD释放71.4%,PBT释放85.6%。PBT释药速度明显快,而PBT中泊洛沙姆含量仅为0.05%,说明吸附技术显著改善了药物的溶出行为。
Tets束谈维负uya等应用水溶性载体PEO及羟丙基纤维素(HPC)分别制备了氟比洛芬(FP)固体分散体。研究表明:FP-PEO固体分散体的释药速度大朝见保伯化乱破节别否攻于FP-HPC固体分散体。在FP-PEO固体分散体中,释药速度随PEO比例的增大而增大。因为FP与PEO可形成氢键,PEO越多,氢键就越多,所以释药细派速度也越快。
Barker等试将液态的维生素E制成固体剂型,用单务哥准硬脂酸甘油酯(Gelucire)44/14(医源沿河析掉初熔点44℃)以熔融法(60℃水浴)制备维生素E的SD,含药量可达50%w/w),药物吸收比普通制剂增末止阶所根圆全沙加两倍,提高了生物且力右灯袁外冲工义造难利用度。且维生素E的SD装入胶囊后储存18个月未见维生素E渗出。Manish等用Gelucire50/13(熔点47℃~53℃)作载体制备萘普生、17-酮甾类、消炎痛、睾丸激素、非那西丁、黄体酮等固体分散体,添加硅酸镁铝作表面吸附剂。固体分散体中药物与硅酸镁铝生成氢键而保持无定形态,加速了药物的溶出。
Anne等用超临界流体法制备了吡啶甲磺酸类药物甘露醇SD(共沉淀物)。DSC、FTIR分析显示,药物的胺基与甘露醇的羟基形成氢键,药物以无定形态存在,故加快了药物的溶出。Okonogi等用溶剂法以甘露醇和尿素制备了氧氟沙星SD。X射线衍射法显示,氧氟沙星-尿素SD(1:4)的药物衍射峰显著降低,表明有晶型药物存在;氧氟沙星-尿素SD(1:19)中药物衍射峰消失,表明药物均呈无定形态存在;氧氟沙星-甘露醇SD(1:19)仍有药物衍射峰,表明也有晶型药物存在。这些提示,作为氧氟沙星SD的载体,尿素优于甘露醇。氧氟沙星SD的溶出曲线表明:甘露醇SD未能显著增加药物的溶出度,而尿素则较大地增加了药物的溶出,证实了尿素的效果好。
MGK是将天然树胶粉碎(100目)经120℃热处理两小时而制得的。其优点是黏性降低,从1800厘泊降至550厘泊。Murali等采用研磨法制备尼莫地平-MGK固体分散体(1:9),药物溶解速度有显著改善,无需加入有机溶剂或高温制备;而且随MGK用量增加,尼莫地平的溶解速度也增加。
■胶原蛋白水解产物
Renata等以胶原蛋白的酶水解产物Gelitacollagel(KLH,分子量18300)作SD载体,用喷雾干燥法制备奥沙西泮SD。X射线衍射法显示,不同载体用量的SD中奥沙西泮衍射峰均消失,表明药物均呈无定形态。SD中奥沙西泮4小时药物溶出27.8%~29.1%,比原料药(5%)显著提高。
固体分散体按药剂学释药性能分为速释型固体分散体,缓(控)型固体分散体和靶向释药型固体分散体。
速释型固体分散体就是利用强亲水性载体制备的固体分散体系,这种类型的固体分散物在固体分散体研究中占绝大比重。
对于难溶性药物而言。利用水溶性载体制备的固体分散物,不仅可以保持药物的高度分散状态,而且对药物具有良好的润湿性。这在提高药物溶解度,加快药物溶出速度,从而提高药物的生物利用度方面具有重要的意义,例如西南制药三厂用熔融法,以PEG6000为载体,制成灰黄霉素滴丸,结果表明,固体分散物口服2h内几乎完全吸收,而微粉片30-80h内方吸收44.3%,药物-载体比1:10-1:5的灰黄霉素分散物在人体内的吸收量比微粉片高1倍多。
速释型固体分散体所用的载体多为高分子化合物,有机酸及糖类,主要有聚乙二醇(PEG)4000和6000、聚乙烯吡咯烷酮(PVP)、尿素、枸橼酸、琥珀酸、去氧胆水、甘露醇、木糖醇、山梨醇、半乳糖等,对水溶性固体分散载体的研究出现了由单一载体向联合载体及加表面活性剂的载体方向发展趋势[1]。
缓(控)释型固体分散体是指以水不溶性或脂溶性载体制备的固体分散体,此分散系可以看作溶解扩散或骨架扩散体系,释放机理与相应的缓释制剂和控释制剂相同,有一级过程,Higuchi过程和零级过程。Nagib N.等人以乙基纤维素为载体,用溶剂法制备了磺胺嘧啶的固体分散体,体外溶出试验结果表明,该固体分散体释药过程的动力学是表观零级式和控制扩散,M.P. Oth等人研究发现,以Eugragit RS 和RL为载体,制备的吲哚美辛-Eugragit共蒸发物体外释药过程符合Hifuchi's时间平方根模型。
缓(控)释型固体分散常用的载体主要有乙基纤维素、蜡脂、Eugragit等。
肠溶型固体分散就是利用肠溶性材料为载体,制备的靶向于肠道溶解释放药物的固体分散体。传统的固体分散体的研究绝大多数都是以水溶性载体如聚乙二醇(PEG)聚乙烯吡咯烷酮(PVP)等促进难涪性药物迅速溶解释放的固体分散体研究,这在促进药物释放和提高生物利用度方面是确有成效的。随着药剂学的发展和新辅料的出现,已经逐渐出现了一些肠溶固体分散体的研究,例如硝苯吡啶肠溶固体分散体的研究,硝苯吡啶为水难溶性药物,生物利用度低,Haswgawa将硝苯吡啶与以乙醇-氯甲烷混合溶剂溶解后,喷雾在蔗糖表面上,制成肠溶固体分散物,体外溶出试验表明,该固体分散物在胃液中溶出极少(50min内少于0.4mg/L)。而在PH5.8的肠液中释放却大大加快(30mmin时达到60mg/L);动物(狗)体内实验表明,该肠溶固体分散体的生物利用度与硝苯吡啶-PVP共沉淀物的生物利用度相近,而且有效血药浓度维持时间前者较后者长,而硝苯吡啶结晶粉末的生物利用度只有肠溶固体分散体的17%。进一步用HP-55硝苯吡啶肠溶固体分散体与欧洲市售品缓释片相比较,发现含药量相当于l0mg的硝苯吡啶HP-55固体分散体颗粒剂与含药20mg的硝苯吡啶缓释片几乎显示出相同的血药浓度特征曲线,因此,该固体分散体颗粒剂可以说是一种吸收率高的缓释制剂。地高辛肠溶固体分散体和潘生丁肠溶固体分散体也显示了同样的结果。
可见,利用肠溶性材料制成的固体分散体,能够使许多难溶性药物的生物利用度提高,而且具有缓释性,这在解决以往利用控制溶解制备水难溶性药物的缓释制剂生物利用区较差的问题是一个很有益的启发。
肠溶性固体分散体常用的载体有:羟丙基甲基纤维素邻苯二甲酸酯(HP-55),醋酸纤维素邻苯二甲酸酯(CAP),Ⅱ、III号丙烯酸树脂,Eugragit L 100和S100,羧甲基乙基纤维素(CMEC)等。
载体材料的性质对固体分散体的性质有很大影响,载体材料应该具有无毒、无致癌性、不影响药物稳定性、不与药物发生化学变化、不影响药物的药效与含量监测等基本性质。
常用的固体分散体载体材料可以分为三大类:水溶性载体材料,包括聚乙二醇(PEG)、聚维酮(PVP)、表面活性剂、有机酸、糖与醇等;难溶性载体材料,包括纤维素、聚丙烯酸树脂等;肠溶性载体材料,包括纤维素类和聚丙烯酸树脂类。
固体分散体的制备常用的方法有熔融法、溶剂法、溶剂-熔融法、溶剂喷雾冷冻干燥法、研磨法等。
1)载体使药物处在高度分散状态。
2)强亲水性载体可增加难溶性药物的溶解度和溶出速率,从而提高药物的生物利用度;难溶性载体可延缓或控制药物释放;肠溶性载体可控制药物于小肠释放。
3)利用载体的包蔽作用,可延缓药物的水解和氧化。
4)载体可掩盖药物的不良气味和刺激性。
5)使液体药物固体化。
6)药物分散状态的稳定性不高,久贮易产生老化现象。
7)滴丸为固体分散体,基质和冷却剂的种类还有限。
 关注微信
关注微信