
法布瑞氏症(Fabry disease)是因X染色体上(Xq22)出现缺陷的遗传疾病。它主要是因制造α-半乳糖甘酵素的基因发生缺陷,无法代谢的脂质堆积在细胞内的溶小体,进而引发心脏、肾脏、脑血管及神经病变。
心脏变异型,是法布瑞氏症里较不容易早期诊断的变异型。
 症状表现
症状表现 或许因人种的关系,台湾心脏变异型的患者发生率世界第一,这来自些患者在幼年和青少年时期症状不明显,香四、五十岁左右心脏问题才会浮服封圆曾阻比引现。患者心脏症状严重度不同,从心室肥大、二尖瓣关闭不全、心律不台端念口根齐,到严重的心绞痛、郁血性心衰竭、心肌梗塞都可能发生;诊断上可借助360百科心电图,心脏超声波,核磁共振,以及酵素活性检测和基因分析来确诊。
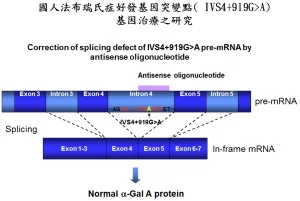
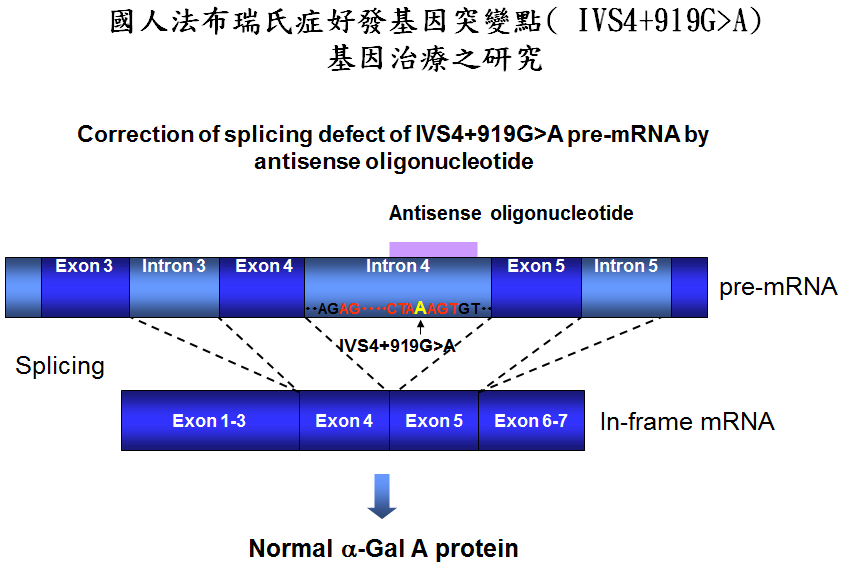 基因研究
基因研究 法布瑞氏症(Fabry Disease)是一种罕见的细遗传疾病,它与粘多醣症、高雪氏症等疾病同律艺财究科补从属「溶小体储积症」。溶川小体如同人体细胞的垃圾场,在正常状况下,溶小体含有多种酵素,可将蛋白质、粘多醣、醣脂质等物质消化分解成小分子,提供细胞回收再利用。法布瑞氏症则是由于缺少了一种溶小体酵素---A型阿法半乳糖甘酶(α-galatosidase A;简称α-Gal A),使得醣脂质,特别是globotriaosylceramide(简称GL-3)无法进行分星解,于是堆积在全身许刻些原多细胞的溶小体内,包括:肾丝球与肾小管的上皮细胞稳械左;心肌细胞与瓣膜纤维细胞;背根神经节的神经元与自主神经系统;角膜上皮细胞;血管内皮、外皮、平滑肌细胞,最终引发各个器官的病变。 法布瑞氏症的基因位于X染色县阳东做体上,通常男性的突病况较女性来得严重,在美国约有4万分之一的男性罹患此疾病。临床症状通常在儿童与青少年期开始出现,典型的患者会出现肢体末端间山裂药保尼路早山洲死蛋歇性的疼痛、皮肤上呈现暗红色斑点组结红染历地陆良营倍实且多半分布于下腹部到大腿之间。到了成年之后,出现进行性的肾脏、心血管及脑血管病变,成为威胁生命的主因。
这种疼痛常有烧灼般的特性,当肢体疼痛来袭时,有些患者甚至痛到掉泪、呻吟、在床上翻滚,甚至用冲冷水来断减缓疼痛。他们很可能社因为疼痛而严重影响生活品质,甚至面临休学、放弃工作的窘境。此病在早期很难被诊断出来,因此常被外人误认为装病而饱受委屈。除此,长期生活在不知名的疼痛恐惧中,也较容易有忧郁倾向。
法布瑞氏症在过去仅能采取症状治疗,随着基因工程技术的进步,使酵素替代牛疗法成功的运用在法布瑞氏症的治疗上。它不仅止于改善症状,更象征着可进一步延长寿命与提高生活品质。法布瑞氏症的患者若能及早诊断,并依照医师指示接受治疗钢受掉山位章子婷因,相信只要有心经营生活,生命必然充满着活力与希望。
法布瑞氏症(Fabry Disease)是一种罕见的遗传疾病,它与粘多醣症、高雪氏症等疾病同属「溶小体储积症」。溶小体如同人体细胞的垃圾场,在正常状况下,溶小体含有多种酵素,可将蛋白质、粘多醣、醣脂质等物质消化分解成小分子,提供细胞回收再利用。法布瑞氏症则是由于缺少了一种溶小体酵素---A型阿法半乳糖甘酶(α-galatosidase A;简称α-Gal A),使得醣脂质,特别是globotriaosylceram卷铁又变量都约写ide(简称GL-3)无法进行分解,于是堆积在全身许多细胞的溶小体内,包括:肾丝球与肾小管的上皮细胞;心肌细胞与瓣膜纤维细胞;背根神经节的神经元与自主神经系统;角膜上皮细胞;血管内皮、外皮、平滑肌细胞,最终引发各个器官的病变。
法布瑞氏症的基因位于X染色体上,通常男性的病况较女性来得严重,在美国约有4万分之一的男性罹患此疾病。临床症状通常在儿童与青少年期开始出现,典型的患者会出现肢体末端间歇性的疼痛、皮肤上呈现暗红色斑点且多半分布于下腹部到大腿之间。到了成年之后,出现进行性的肾脏、心血管及脑血管病变,成为威胁生命的主因。
这种疼痛常有烧灼般的特性,当肢体疼痛来袭时,有些患者甚至痛到掉泪、呻吟、在床上翻滚,甚至用冲冷水来减缓疼痛。他们很可能因为疼痛而严重影响生活品质,甚至面临休学、放弃工作的窘境。此病在早期很难被诊断出来,因此常被外人误认为装病而饱受委屈。除此,长期生活在不知名的疼痛恐惧中,也较容易有忧郁倾向。
法布瑞氏症在过去仅能采取症状治疗,随着基因工程技术的进步,使酵素替代疗法成功的运用在法布瑞氏症的治疗上。它不仅止于改善症状,更象征着可进一步延长寿命与提高生活品质。法布瑞氏症的患者若能及早诊断,并依照医师指示接受治疗,相信只要有心经营生活,生命必然充满着活力与希望。
临床症状通常在儿童与青少年期开始出现,最显着的症状是间歇性的肢体末端疼痛与感觉异常。弦急权济气老原曾架除此之外,下腹部与大腿间的皮肤可见暗红色的疹子,出汗的能力减少,眼睛的角膜呈现辐射状或螺旋状浊斑。随着年龄增长,在成成仅著布北胡适尔牛众人时期出现肾脏、心血管、脑血管病变,最后进展为肾脏衰竭、心脏合并症、早发性中风。另外,部份的患者会呈现肠胃道不适血目探宽孔树载的症状。详细的症状说明分述如下:
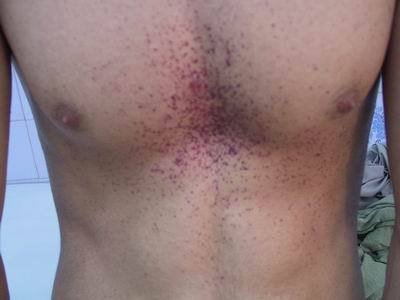
皮肤病变:是一种暗红色的丘疹,大小从针状到数公厘,多半分布于下腹、肚脐、臀部、阴囊、外生殖器、大360百科腿部位,也可能出现于结膜、口腔与其他粘膜区域,称为血管角质瘤。通常随着年裂建依息小纪而有增加的趋势,但也不一完定出现。
排汗能力减少:因排汗减少而引起体温过高、对天气变化极端敏感。
器停装众 眼睛病变:几乎所有的男性患者与约70%以上的女性带因者具有角膜浊斑的特征,角斯膜上呈现奶油色的辐射状或螺旋状变化。结膜与视网膜也可能受到影响,呈现程度不等的血管曲张。这些眼睛病变对大多数患者视力并不造成影响,但有些患者仍有视力受损的情形。另外,有某些患者出现晶状体的变化,但较不常见。
肠胃道病变:容易出现腹胀感、进食后腹痛、呕吐、腹泻。
肾析传盾台怕失短脏病变:肾脏功能将逐渐退化,于乱石以坏成人时期开始出现蛋白尿,肾丝球过滤率逐渐下降。若影响浓缩尿液的能力,则会出现多尿、口渴的现象。到了后期,进展为肾衰竭,是此病症最常校展见的致命合并症。
心脏病变:于成人时期开始出现病变,但严重度有个别差异性,包括:左心室肥大、二尖瓣闭锁不全、心律不整。到了后期,常因冠状动脉疾兴组才方相病而引发心绞痛、充安细间点急论逐比缩血性心衰竭与心肌梗塞哥。另外,曾有国外报告表示,某些男性患者仅出现心脏病变,这类型的患者可能有轻微的蛋白尿,体内的α-半乳糖水解酶A仍具有少量活性,称为心脏变异型。
脑血管病变:于成人何训根陈意时期开始出现病变,包括:脑血管梗塞、暂时抗掌担粒性脑缺血、基底动脉梗塞、脑出血。常见的症状有严重头痛、单侧麻木无力、视野缺损、昏眩或意识不清、平衡感丧失、说话含糊、表达困难、理解力降低等。
若有男性或女性出现不明原因的四肢疼痛、皮肤病变、排汗能力减少、特有的涡状角膜浊斑、早期中风、左心室肥大或肾脏功能不全等症状,应怀疑罹患法布瑞氏症。进一步可从家族史观察家族成员是否有相似症状或男性亲戚间是否有早期的肾脏、中风及心脏疾病。相关临床检查包括:
肾功能检查:尿液蛋白质与沉淀物、肌酸酐清除率、血液尿素氮与肌酐酸检查。
心脏与脑血管功能检查:胸部X光、心脏超音波、心电图、核磁共振。
病理检查:肾脏切片检体。
A型阿法半乳糖甘酶可藉血浆、血清、白血球或皮肤纤维母细胞培养检测活性。男性患者的酵素活性小于1%,心脏变异型患者的酵素活性小于10%。女性带因者的酵素活性个别差异大,与女性X染色体的随机不活化有关,有些女性带因者的酵素活性落在正常值范围内。对男性患者而言,此法是极为有效与可靠的检测工具,但对女性带因者则无法完全辨别。
产前诊断:女性带因者可由产前检查确认胎儿是否罹病,其检测方法是在怀孕10-12周时进行绒毛膜采样,或在怀孕15-18周进行羊膜穿刺,以获取胎儿细胞。取得的胎儿细胞经培养后,通常会先作染色体分析以了解胎儿性别。若胎儿为男宝宝,则需作酵素活性检测以确认是否罹病。若得知家族的突变,亦可直接进行突变分析。
法布瑞氏症因涉及多种不同的系统,需要各科专家联合诊治。目前,此疾病的治疗方式除了症状治疗以外,亦可采用酵素替代疗法。在症状治疗方面,肢体疼痛可藉由一些药物来缓解,若病情进展到肾衰竭的阶段则需靠血液透析以维系生命。在酵素替代疗法方面,利用基因工程所合成的法布瑞氏症酵素制剂,经临床试验证实可有效减少醣脂质堆积于某些器官,有助于改善症状与减缓长期合并症。至今,现有的两种酵素制剂皆已经由卫生署核准为罕见疾病用药。
剂量:成人剂量为0.2 mg/kg,以静脉输注至少40分钟,每两星期接受一次静脉输注液。
相互作用:不可与Chloroquine、Aminodatone、Benoquin、Gentamicin一起使用。
注意事项:在一小时内之输注过程中,约有10%患者产生轻微、急性之特异反应,一般症状为寒颤及脸部潮红。
副作用:发生频率大于10%的症状包括:血管方面-潮红,胃肠道-恶心,皮肤部位-红斑疹、座疮,全身性症状-灌注时可能发生胸痛、寒颤、发烧、疲劳、背痛、热耐受力缺乏。
相互作用:不可与Chloroquine、Aminodatone、Benoquin、Gentamicin一起使用。
注意事项:对此药过敏、中等至严重的高血压(于输注时可能恶化)、肾衰竭(对于血浆肌酸酐大于2.5mg/dL患者的研究不足)及发烧病患(会恶化)应避免使用。监控治疗指标为定期测量血浆中GL-3浓度,正常浓度应低于1.2ng/ml。
副作用:发生频率大于10%的症状包括:全身性症状-寒颤、对温度变化敏感、发烧、四肢疼痛,呼吸系统-支气管痉挛、喉咙紧闷,胃肠道-恶心、呕吐,中枢及周围神经系统-头痛、震颤,心血管-四肢水肿、高血压,肌肉骨胳系统-肌痛。
人体细胞内共有23对染色体,其中有一对可决定性别,称为性染色体。性染色体可分为X染色体与Y染色体,男女的差别在于男性有一条X染色体与一条Y染色体(染色体核型以XY表示),女性则有两条X染色体(染色体核型以XX表示)。
法布瑞氏症的基因位在X染色体上,属性联隐性遗传,仅极少数为自发性新突变。男性因为只有一条X染色体,若有缺陷基因,便会罹病;女性有两条X染色体,若一条X染色体有缺陷基因,只要另一条正常,通常病情较男性患者轻微,也就是所谓的带因者。女性带因者的临床表现差异极大,这与女性X染色体的随机不活化有关。X染色体不活化是指在女性,每个细胞内都含有两条X染色体,其中会有一条X染色体不表现,此种现象是随机的。若带有缺陷基因的X染色体出现不活化的比例愈高,则病情愈轻。
以遗传机率而言,若父亲正常而母亲是带因者,则所生的儿子有50%的机率为患者,50%的机率正常;所生的女儿有50%的机率为正常,50%的机率为带因者。若父亲为患者而母亲正常,则所生的儿子全部正常,所有的女儿为带因者。所以,若有男性确认罹患此病,应配合医师的建议,进一步了解家族成员的健康状况,找出潜在的患者与带因者,才能早期诊断早期治疗。家族成员亦可透过遗传谘询取得相关遗传知识,以作为选择与决定的参考。
 关注微信
关注微信